New preprint announcement
Ab initio thermodynamic statistical modeling of the miscibility gap and the metal–insulator transition in SrTi1-xVxO3
We are pleased to announce the submission of our latest preprint to arXiv, titled Ab initio thermodynamic statistical modeling of the miscibility gap and the metal–insulator transition in SrTi1-xVxO3.
SrTiO3 is a textbook band insulator, while SrVO3 is a moderately correlated metal. Substituting vanadium for titanium in the solid solution SrTi1-xVxO3 therefore offers a clean knob to tune a material continuously between the two, and experiments indeed see a composition-driven metal–insulator transition (MIT). The details, however, have stubbornly resisted a clean theoretical picture: reported critical concentrations scatter across xc ≈ 0.4–0.7, and the mechanism sits at the complicated intersection of Mott physics and disorder-induced (Anderson) localization.
Part of the problem is methodological. Previous first-principles studies had to commit to a single supercell at each composition. But at a given x there are many symmetry-inequivalent arrangements of V and Ti on the cation lattice, and they can have markedly different electronic properties. A real, thermally disordered solid solution is not any one of these configurations—it is an ensemble of many of them, populated according to temperature. Picking one cell, even the lowest-energy one, quietly throws that physics away.
In this work, we tackle the alloy within the generalized quasichemical approximation (GQCA), a thermodynamically consistent framework in which every quantity is computed as an ensemble average over all symmetry-inequivalent clusters, weighted by occurrence probabilities that minimize the Gibbs mixing free energy. This gives a well-defined recipe for averaging over supercells and puts the structural and electronic descriptions on an equal footing.
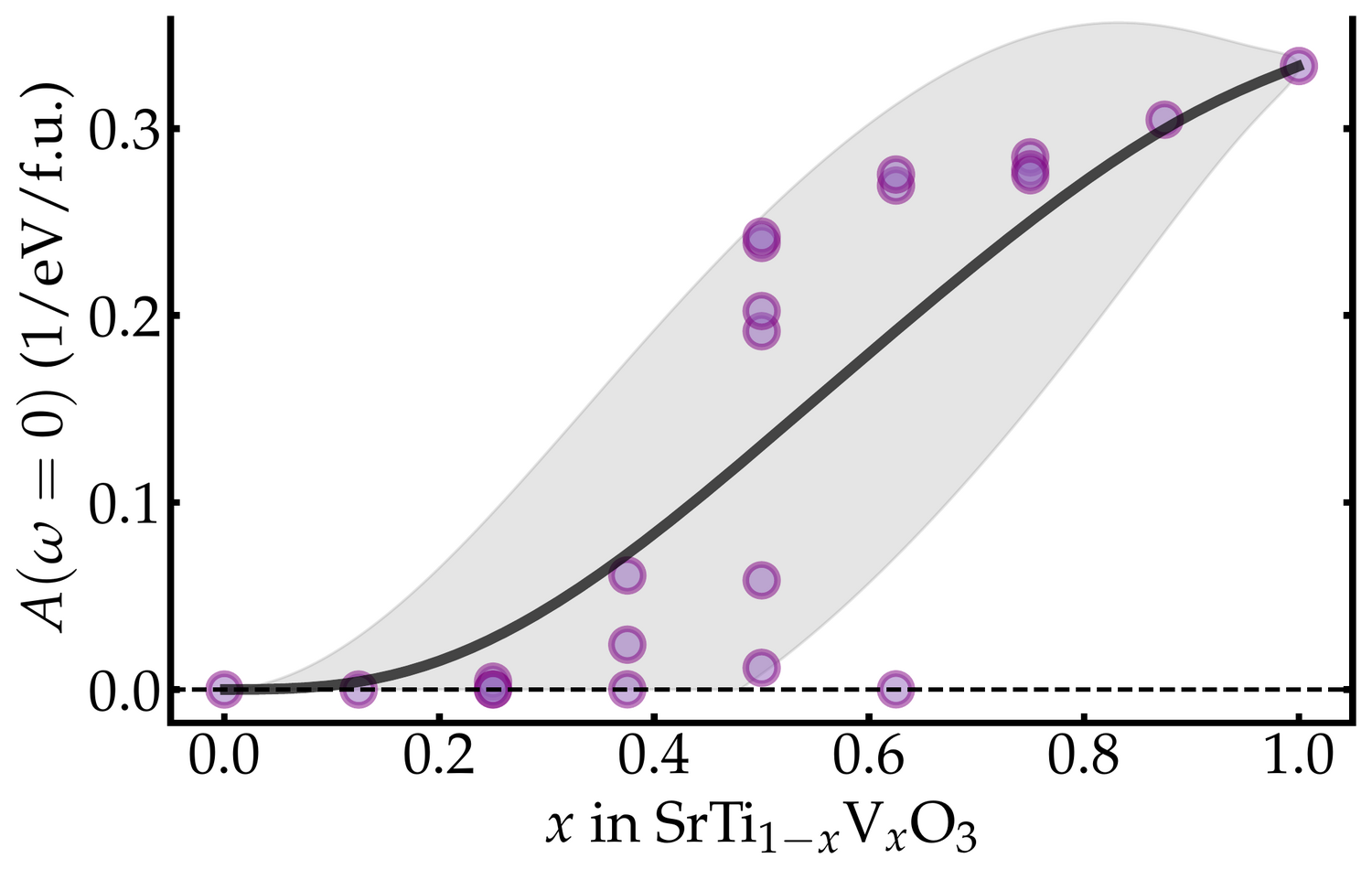
From the mixing thermodynamics we obtain a miscibility gap with a critical temperature of 1443 K, consistent with the limited experimental evidence for high-temperature solubility and subsequent decomposition, and substantially larger than earlier cluster-expansion estimates. Coupling the cluster ensemble to dynamical mean-field theory (DMFT) then lets us track the density of states at the Fermi level across the whole composition range. The contrast is striking: plain density-functional theory predicts a metal for every x > 0, whereas the correlated spectral function reproduces the transition, evolving from insulating below x ≈ 0.3 to metallic near x = 1. Finally, classifying each cluster as metallic or insulating and running site percolation on a simple cubic lattice yields a sharp onset of system-spanning conduction near x ≈ 0.4.
Together these results provide a unified, ab initio picture of the MIT in SrTi1-xVxO3, and—perhaps more importantly—a thermodynamically consistent, configuration-averaged framework that should carry over to the broader class of correlated materials in which substitutional disorder governs, or even drives, electronic transitions.
We’re excited to share this work and look forward to the community’s feedback!
Check out the preprint arXiv:2607.07067.